Antibody-drug conjugates (ADCs) are currently at the forefront of the drug development revolution occurring in oncology delivering on the long-standing promise of a highly effective and selective cancer treatment. They are among the fastest-growing drug classes in oncology with a remarkable surge in marketing approvals in recent years. Nine ADCs were FDA approved from 2017 onwards, and 14 in total for both hematologic malignancies and solid tumors (see Table 1). Daiichi Sankyo and AstraZeneca are leading the way with the blockbuster anti-Her2 Enhertu indicated for unresectable or metastatic HER2-positive solid tumors and soon to be moving into first-line treatment. Moreover, an impressive clinical pipeline with over 100 candidates is in place. The many recent deals and partnerships in the ADC arena suggest that there is continued significant interest from large pharma, also evidenced by the recent multi-billion acquisitions of Seagen by Pfizer and ProfoundBio by GenMab.
Table 1. Overview of antibody-drug conjugates for Hematological malignancies and solid tumors. Table adapted from Fu et al. 2022 Antibody drug conjugate: the “biologic missile” for targeted cancer therapy, Signal Transduction and Targeted Therapy 7, Art no.: 93.[External Link]
| ADC name (Company) |
Trade Name | Target Antigen |
Linkers | Linker Type | Payloads | Payload Type | Average DAR | Approval Date | Indication |
|---|---|---|---|---|---|---|---|---|---|
Hematological malignancies |
|||||||||
| Gemtuzumab ozogamicin (Pfizer) |
Mylotarg® | CD33 | hydrazone | Acid sensitive, cleavable |
N-acetyl-γ- calicheamicin |
DNA damaging | 2-3 | 2000 & 2017 | AML |
| Brentuximab vedotin (Seagen) |
Adcetris® | CD30 | mc-VC-PABC | Peptidase sensitive, cleavable |
MMAE | Tubulin inhibitor | 4 | 2011 | HL, ALCL, PTCL, cHL |
| Inotuzumab ozogamicin (Pfizer) |
Besponsa® | CD22 | hydrazone | Acid sensitive, cleavable |
N-acetyl-γ- calicheamicin |
DNA damaging | 5-7 | 2017 | B-cell precursor |
| Moxetumomab pasudotox (AstraZeneca) |
Lumoxiti® | CD22 | mc-VC-PABC | Peptidase sensitive cleavable |
PE38 | Translation inhibitor | N/A | 2018 | HCL |
| Polatuzumab vedotin (Roche) |
Polivy® | CD79B | mc-VC-PABC | Peptidase sensitive, cleavable |
MMAE | Tubulin inhibitor | 3.5 | 2019 | DLBCL |
| Belantamab mafodotin (GSK) |
Blenrep® | BCMA | mc | non-cleavable | MMAF | Tubulin inhibitor | 4 | 2020 | MM |
| Loncastuximab tesirine (ADC Therapeutics) |
Zynlonta® | CD19 | dipeptide | Peptidase sensitive, cleavable |
PBD dimer | DNA crosslinking | 2.3 | 2021 | Large B-cell lymphoma, DLBCL |
Solid tumors |
|||||||||
| Ado-trastuzumab emtansine (Roche) |
Kadcyla® | HER2 | SMCC | Non-cleavable | DM1 | Tubulin inhibitor | 3.5 | 2013 | HER2-positive early breast cancer |
| Enfortumab vedotin (Seagen) |
Padcev® | Nectin-4 | mc-VC-PABC | Peptidase sensitive, cleavable |
MMAE | Tubulin inhibitor | 3.8 | 2019 |
Urothelial cancer
|
| Fam-trastuzumab deruxtecan (Daiichi Sankyo) |
Enhertu® | HER2 | tetrapeptide | Peptidase sensitive, cleavable * |
DXd | DNA topoisomerase I inhibitor |
7-8 | 2019 | HER2- positive breast cancer |
| Sacituzumab govitecan (Immunomedics) |
Trodelvy® | Trop-2 | CL2A | Peptidase sensitive, cleavable * |
SN38 | DNA topoisomerase I inhibitor |
7.6 | 2020 | TNBC |
| Cetuximab sarotalocan (Rakuten Medical)** |
Akalux® | EGFR | N/A | N/A | IRDye700DX | Membrane disruption | 1.3-3.8 | 2020 | HNSCC |
| Disitamab vedotin (RemeGen) |
Aidixi® | HER2 | mc-VC-PABC | Peptidase sensitive, cleavable |
MMAE | Tubulin inhbitor | 4 | 2021 | Gastric cancer |
| Tisotumab vedotin (Genmab/Seagen) |
Tivdak® | TF | mc-VC-PABC | Peptidase sensitive, cleavable |
MMAE | Tubulin inhibitor | 4 | 2021 | Cervical cancer |
DAR Drug-to-Antibody ratio, AML acute myeloid leukemia, mc-VC-PABC maleimidocaproyl-valine-citrulline-p-aminobenzoyloxycarbonyl, MMAE monomethyl auristatin E, MMAF monomethyl auristatin-F, HL Hodgkin lymphoma, ALCL anaplastic large cell lymphoma, SMCC succinimidyl‐4‐(N‐maleimidomethyl)cyclohexane‐1‐carboxylate, DM1 derivative of maytansine 1, HER2 human epidermal growth factor receptor 2, cHL classical Hodgkin lymphoma, PTCL peripheral T-cell lymphomas, MM multiple myeloma, PE38 a 38kD fragment of Pseudomonas exotoxin A, DLBCL diffuse large B-cell lymphoma, DXd Exatecan derivative for ADC, CL2A a cleavable complicated PEG8- and triazole-containing PABC-peptide-mc linker, SN38 active metabolite of irinotecan, HCL hairy cell leukemia, TNBC triple-negative breast cancer, HNSCC head and neck squamous cell carcinoma, BCMA B-cell maturation antigen, EGFR epidermal growth factor receptor, GSK GlaxoSmithKline Inc., PBD pyrrolobenzodiazepine, TF tissue factor, mc maleimidocaproyl.
* High DAR, site directed conjugation, ** Payload requires localized application of near-infrared light for activation
The Components of ADCs
The ADC concept is appealing because it potentially combines a high target specificity with potent tumor cytotoxicity. The elegancy of this approach is that key strengths of biological and chemical therapeutics are combined in one molecule and should thereby overcome the limitations of conventional cancer chemotherapy improving the therapeutic window and reducing off-target side effects. As shown in See Figure 1, ADCs are typically composed of three structural elements: a payload (1) attached to a monoclonal antibody (2) via a linker (3). Each of these structural elements including conjugation technology and stoichiometry of payload must be optimized to achieve stability in the bloodstream, accurately and sufficiently reach the target cancer cells, and ultimately release the cytotoxic payload in close proximity to the specified cancer cells for effective treatment.
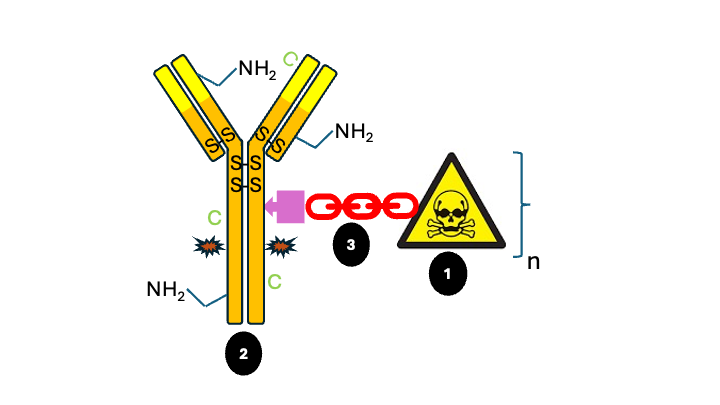
Figure 1: Schematic representation antibody-drug conjugate: variable region, target binding site; S-S; native interchain cysteine bridge, C; cysteine introduced by site-directed mutation at C-terminus light chain, NH2 and C; examples of native lysine and cysteine residues open for stochastic linker payload conjugation; * glycosylation site N297, n; a variable number of linker payloads can be conjugated to the antibody. See text for further explanation.
The Mode of Action of ADCs
All marketed ADCs have a highly similar Mode of Action (see Figure 2). After intravenous administration and tissue distribution, the antibody moiety of the ADC binds to the tumor antigen (step 1) upon which the ADC is internalized by endocytosis (steps 2 & 3). In the endosome, the ADC is degraded resulting in the release of the payload (step 4). The activated payload diffuses to the cytosol and nucleus and binds to DNA or microtubules, depending on the payload (step 5), thereby triggering cell death by apoptosis (step 6). In addition, the activated payload can diffuse into the tumor microenvironment and kill (tumor) neighbor cells including antigen-negative tumor cells (so-called bystander effect), an effect which may be especially beneficial in solid tumors. In addition, the anticancer activity of ADC may also involve antibody-dependent cellular toxicity & phagocytosis, and complement-dependent cytotoxicity via the Fc segment on the antibody binding to Fc receptor on innate immune effector cells like NK cells and macrophages.
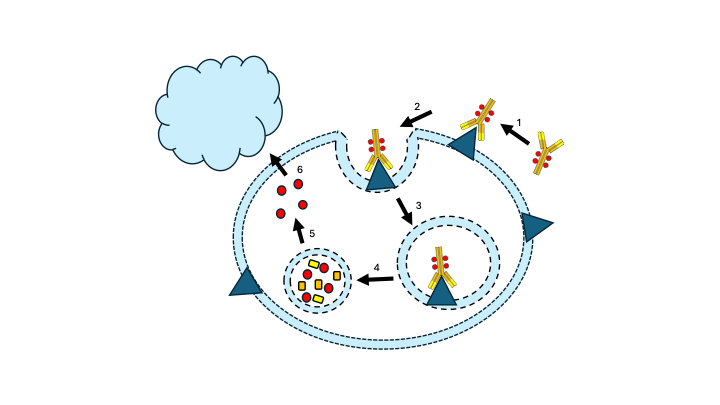
Figure 2. The general mechanism of action for an ADC. See text for explanation.
Early Days of ADCs
The successful development of chemotherapy in the 1940s and monoclonals in the 1970s allowed for the development of the first ADCs in the 1980s. It took nevertheless decades of optimization of all ADC structural elements to reach clinical success in the form of market approval, leaving behind a high number of clinical failures. The first ADC consisted of the low-potent conventional chemotherapy drug doxorubicin conjugated via an unstable, non-cleavable, acid-sensitive hydrazone linker to a chimeric, mouse derived antibody. The result was an immunogenic molecule with off-target safety issues with low potency: no surprise that basically all of the design aspects of this first ADC have been aborted. Over the years, the mAbs, the conjugation technology, the linker design, and the payload were all dramatically improved. This has resulted in a wide choice of available structural elements potentially leading to an immense diversity in lead candidates, all with different pharmacological and clinical properties.
ADC Linkers
The first major improvement was the use of humanized antibodies together with higher potency payloads, thereby greatly improving efficacy and lowering immunogenicity. This approach resulted in the first marketed product (see Table 1). However, the use of acid-labile linkers often resulted in premature release of the payload in circulation followed by off-target toxicity, emphasizing the need for more stable linkers. These first-generation linkers were often conjugated stochastically to native free lysines or reduced cysteines via an active ester or maleimide reagent, respectively, which are unfortunately unstable in the bloodstream due to the retro-Michael reaction.
Recent marketed ADCs use more stable maleimide conjugation chemistry, for example, by using maleimidocaproyl conjugated to native reduced cysteines, thereby lowering off-site toxicity significantly. To exert its activity, the payload needs to be released from the linker-antibody moiety. The manner in which the drug is released from the ADC depends on the type of linker used. They can basically be functionally divided into cleavable and non-cleavable types. The cleavable linker variants generally use either the low pH in endosomes/lysosomes after endocytosis (example Table 1) or proteolytic cleavage through the highly tumor-expressed Cathepsin B protease to release the payload. ADCs with non-cleavable linkers depend on endocytosis-mediated proteolysis for the release of the payload.
Uncleavable linkers generally have higher plasma stability, resulting in lower off-target toxicity. As a result of charged amino acid appendages caused by incomplete proteolysis, the payload often lacks membrane permeability. This lack of permeability severely reduces the payload’s capacity to kill neighboring tumor cells, including those that may not express the relevant antigen on their cell surface or are less accessible directly from the circulatory system. For this reason, the application range of the ADC containing the non-cleavable linker is considered limited, and it is mainly used for the treatment of hematological cancers or tumors with high antigen expression. In contrast, release of the drug via a cleavable linker often results in an uncharged drug with a high membrane permeability, enabling the killing of neighboring tumor cells (so-called bystander effect).
Conjugation
Most marketed ADCs have the payload stochastically conjugated to native cysteines present in the antibody. The cysteines are first reduced, followed by conjugation of the payload using maleimide chemistry. This stochastic approach, where a small fraction of the available cysteines or lysines are used for conjugation, results in a mixture of drug products with a varying drug-to-antibody ratio (DAR). This heterogeneous mixture can affect the therapeutic index, as species with a high DAR may not only be prone to ADC aggregation (due to the increased hydrophobicity of the high payload) but can also show fast hydrophobicity-driven clearance in vivo, often resulting in both reduced safety and efficacy. On the other hand, low to no DAR species may present problems related to drug efficacy, as naked antibodies (or lightly drug-loaded species) compete for limited target antigens with higher potency variants.
Moreover, the stochastic approach may result in conjugation to the antibody variable region via available native lysine and cysteine residues (see Figure 1), thereby hampering binding to the target. To overcome DAR heterogeneity driven by a stochastic conjugation process, considerable effort has recently been put into the development of site-specific conjugation, which should lead to a more homogeneous product with respect to DAR and, consequently, a better safety and efficacy profile.
Recent marketed examples are the blockbuster Enhertu (Daiichi Sanko & AstraZeneca) and Trodalvy (Immunomedics), indicated for HER2-positive breast cancer and triple-negative breast cancer, respectively. Daiichi Sanko and Immunomedics use a process that allows for the specific reduction of all four interchain C=C bonds (see Figure 1), thus facilitating eight binding sites for payload conjugation. This results in a highly homogeneous product with a DAR of 8. To overcome the challenges associated with high DAR (higher toxicity and poor PK profiles), Enhertu and Trodalvy have linker payloads with lower potency and modulated hydrophobicity. Additional features thought to contribute to the success of Enhertu are its short systemic half-life to avoid systemic exposure and bystander effect due to the cleavable linker used and the high membrane permeability of the payload.
Clearly, site-specific conjugation and linker stability can improve the therapeutic window of ADCs. More “chemical” site-specific approaches are currently in clinical development. A promising approach is the use of site-directed mutagenesis to incorporate a cysteine in the C-terminal part of the light chain (see Figure 1; C); a site that proved to be receptive for the conjugation of hydrophobic drug compounds (THIOMABTM technology). Site-specific conjugation of the linker payload using maleimide chemistry results in a highly stable, potent and near-homogeneous ADC. The resulting DAR is relatively low (2), thus requiring the use of more potent payloads.
Another promising development is the use of enzymatic strategies to achieve a more homogeneous product. A clinical example is the GlycoConnect technology, which is based on enzymatic remodelling of the native antibody glycan at N297 followed by metal-free click conjugation of the cytotoxic payload (see Figure 1). This enzymatic approach allows for a more selective and efficient conjugation process, resulting in a highly homologous product with DAR2 or 4 with an extended therapeutic index.
ADC Payload Improvement
Only a small portion (2% max) of the ADCs is estimated to reach the targeted tumor site after intravenous administration. Consequently, this low exposure requires a highly potent payload that can eradicate the tumor. The first-generation ADC-chemotherapy drugs like doxorubicin were active in the higher uM range resulting in sub-optimal efficacy. Currently marketed drugs used for ADCs on the other hand show IC50 values in the nM to pM range. All marketed ADCs now either have potent microtubulin inhibitors or DNA-targeting agents coupled to the antibody. Microtubules are the main component of the cytoskeleton and play a significant role in cell division, particularly during the rapid proliferation of tumor cells, making them interesting targets for anti-tumor drugs. Tubulin inhibitors like the auristatin derivatives monomethyl auristatin E & F are among the favorites, used in at least 5 FDA-approved ADCs with IC50 values in the low nM range. Several DNA damaging agents like duocarmycin (DNA alkylation) and pyrrolobenzodiazepine (DNA crosslinking) even work in the picomolar range. These highly effective compounds may even work with a low DAR for those cells with low antigen expression.
Interestingly, two successful ADCs that recently hit the market have used Topoisomerase inhibitors SN38 (Trodelvy) and DXd (Enhertu) with IC50 values in the high nM range. These less efficacious compounds were most likely chosen to compensate for the high DAR (8) used as compared to the other marketed products (see also see previous section).
Conclusion
ADCs have come a long way since the eighties with an accelerating number hitting the market recently, benefiting tens of thousands of patients. All ADC components have seen major improvements to obtain more efficacious and safe products with an acceptable therapeutic index. The latest improvement is the use of site-directed conjugation instead of the stochastical approach leading to a more homogeneous product which significantly improved its therapeutic index. Many more developments are still in the clinic, hopefully leading to ADCs with greater efficacy, fewer side effects and many more indications also outside the field of oncology. Particularly interesting are the developments with respect to drug and targeting device. Next to the standard antibodies, bi-specifics, peptides, FABs and aptamers are tested as targeting devices. Even more interesting is the variety of payloads under development including radioisotopes, immune-stimulating components and oligonucleotides opening up applications outside oncology.
At 3D-PharmXchange, we have a solid track record in this field and believe that successful development of ADCs requires a multidisciplinary approach ranging from hard core chemistry to synthesize the linker payload, knowledge in the field of biologics to produce the antibody, conjugation experience to connect both to assay expertise to guarantee product quality and integration of this with the adjacent fields of preclinical toxicology and clinical development, to optimally balance safety and efficacy potential. Our CMC team can furthermore perform CDMO selection and management, quality assurance and CMC regulatory support. Overall regulatory support for the program as whole is usually also integrated into this.

Gert Barf, PhD (Senior Consultant CMC Small Molecules)
Get in touch with out
team of CMC experts
